什么是基序?
基序(Motif),是DNA、蛋白质等生物大分子中的保守序列。例如,在反式作用因子的结构中,基序一般指构成任何一种特征序列的基本结构,作为结构域中的亚单元,其功能是体现结构域的多种生物学作用。
MoSDi软件(Motif Statistics and Discovery)
在生物学上,我们经常需要预测一个或几个同源的DNA、RNA、或氨基酸序列的基序。这里主要有两个问题:
- 规定一个可以衡量标准的值(例如:得分score)
- 找到一个得分好的基序(或最优的)
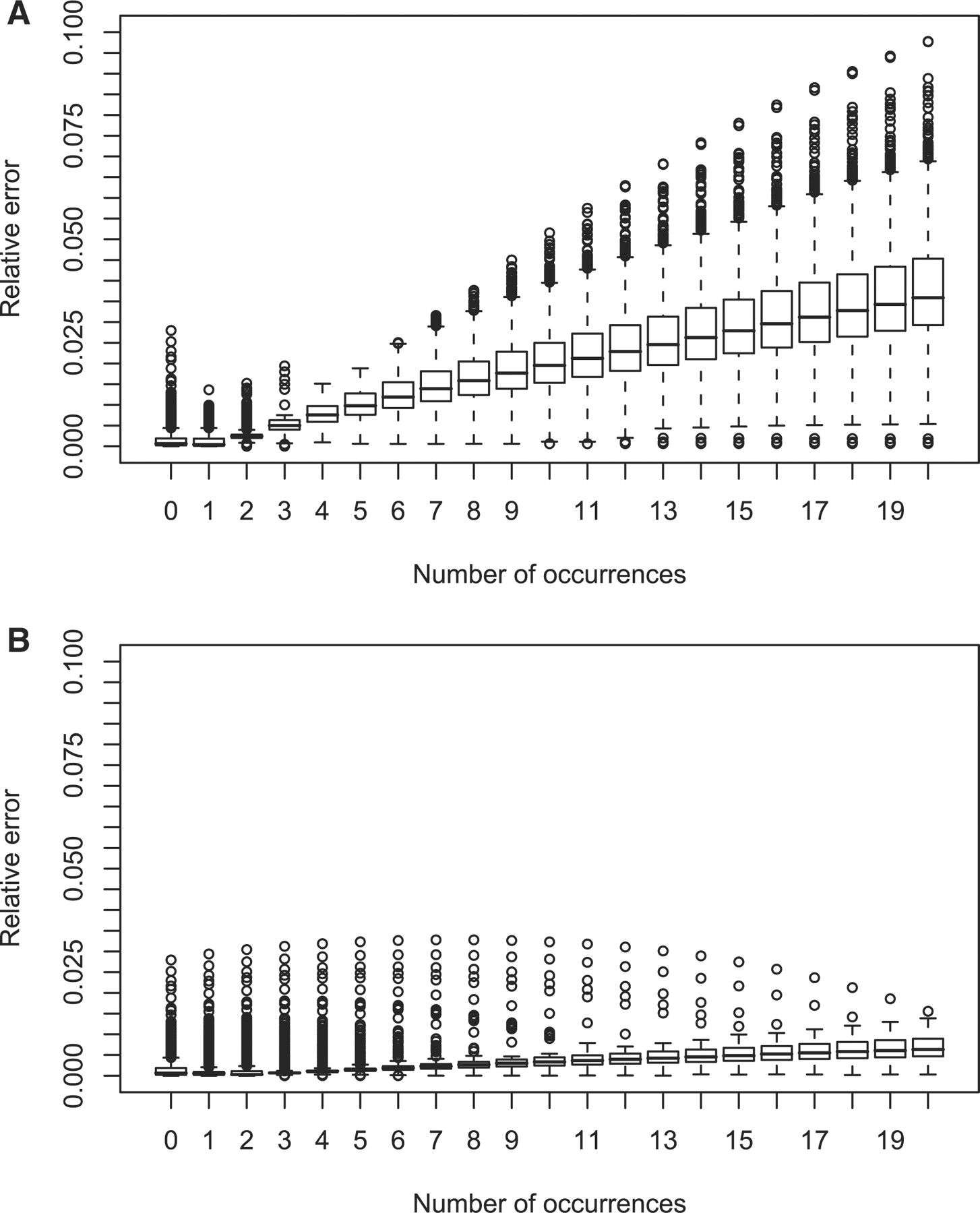
在过去的几年里,通过一些运算法则预测了大量的基序。在我们的研究中,我们试图寻找一种更高效率和更准确的方法来预测基序。最后,我们引入了一种叫probabilistic arithmetic automata的算法,这是一种更快和更准实现基序统计的理论构架(见参考文献1)。在这里,用p值(p-value)来描述预测最优基序的运算法则(见参考文献2)。

MoSDi,是一个基于JAVA的序列基序统计和预测软件。这个软件目前是处在一个试验性的阶段。具有同样预测基因功能的软件有MEME, Weeder。但MoSDi的效果更好。
下载
- MoSDi Revision 468 (as of 2009/01/13): JAR-File
- MoSDi uses jopt-simple to parse command line options. Right now, a version prior to 3.0 must be used (interface is not backwards-compatible and we haven’t ported our code yet). The last 2.x version is 2.4.1.
怎样用MoSDi预测DNA基序
这个方法在Linux下测试有效,在一些类Unix系统上也可用。如果你是用windows,试一下用cygwin搭建一个windows下的类Unix平台。
MoSDi is configured to look for the file jopt-simple.jar in the directory of the MoSDi JAR-file. The easiest way to satisfy this requirement is to create a link named jopt-simple.jar in the same directory as the MoSDi JAR-file:
$ cd <path-to-mosdi-jar> $ ln -s <path-to-jopt-simple>/jopt-simple-<version>.jar jopt-simple.jar
下载这个文件当测试用: this file with (toy-)example sequences. (右键另存为。格式:一行一个序列,目前暂不支持fasta的格式)。出于便利,把这个文件保存在MoSDi JAR-file目录下。
In our toy example, we wish to examine all IUPAC motifs of length 8 with at most 2 of the characters R, Y, W, S, K, M (two-nucleotide-wildcards) and with at most 2 Ns (four-nucleotide-wildcard). At first, we need to generate a list of abelian patterns that satisfy these conditions:
$ java -jar mosdi_r468.jar iupac_abelian_gen -M 8,2,0,2 8 > abelian_patterns
Now we examine the example sequences with respect to these abelian patterns
$ java -jar mosdi_r468.jar discovery -t 1e-15 -F example-sequences abelian_patterns |grep ‘>>’ > results
The switch “-t 1e-15” tells the algorithm to look for motifs with a p-value below 1e-15. Grepping for >> saves us from the software’s rather detailed output. Note that, in order to parallelize the computation, we may split the file abelian_patterns into chunks and process them on different cores/machines.
Sorting results by p-value gives us the winner motif (along with some statistics):
>>p_value>> 3.617957e-37 LIN >>stats>> TAARASGA 1 9 9 17 5.170981e-02 >>poisson>> 5.170981e-02 1.000000e+00 >>runtimes>> 0.000000e+00 0.000000e+00 0.000000e+00
The motif discovery process is as yet restricted to i.i.d. background models. Thus we might want to re-evaluate all returned motifs with respect to a Markovian background model. Firstly, we create a list of all returned motifs:
$ grep '>>p_value>' results | cut -d ' ' -f 5 > motifs
Then, we re-evaluate these motifs w.r.t. an third order Markov model (M3):
$ java -jar mosdi_r468.jar calc_scores -M3 -F example-sequences motifs |grep ‘>>’ > results_M3
详情请看:http://bioinformatics.oxfordjournals.org/cgi/content/abstract/25/12/i356
参考文献
[1] Tobias Marschall and Sven Rahmann. Probabilistic arithmetic automata and their application to pattern matching statistics. In Paolo Ferragina and Gad Landau, editors, Combinatorial Pattern Matching (CPM’08), volume 5029 of LNCS, pages 95-106. Springer, 2008.
[2] Tobias Marschall and Sven Rahmann. Efficient Exact Motif Discovery. Submitted.
Last changed 2009/06/09